Alleviating the Long-term Cardiotoxicity of Cancer Therapies

Kimberly Rockley completed her PhD in the Division of Pharmacy at Durham University before joining us at ApconiX in June last year. Her research utilised in vitro cardiomyocyte models to investigate the link between angiotensin signalling and anthracycline-induced cardiotoxicity. Here she describes the problem of cancer therapy induced cardiotoxicity and the potential impact of her research.
Thesis title: ‘In Vitro Evaluation of Anthracycline-induced Cardiotoxicity and Mitigation by Perturbation of Angiotensin Signalling.’
Alleviating the long-term cardiotoxicity of cancer therapies
The link between angiotensin signalling and anthracycline-induced cardiotoxicity
Although increased understanding of the molecular basis of cancer has advanced treatment and improved cancer survivorship, it is now clear that these new treatments can cause chronic health conditions for survivors. The number of cancer survivors in the UK is estimated to increase by one million each decade from 2010–2040, which equates to three million cancer survivors by 2020 at risk of long-term toxicities.1 Development of therapy-induced cardiovascular toxicity is a potentially life-threatening adverse effect that dramatically reduces quality of life and can be responsible for premature death. Ultimately, today’s cancer patient can become tomorrow’s cardiology patient.2
The scale of the problem
Cardiovascular liabilities occur across the spectrum of cancer therapies, from chemotherapies that interfere with cell division, to the more sophisticated molecularly targeted therapies that perturb intracellular signalling in cancer cells. Toxicities range from acute disturbances such as dysrhythmias and hypertension, to progressive structural changes which can lead to heart failure.3 One such class of cancer therapeutic are the anthracycline class of topoisomerase II inhibiting cytotoxins, which are associated with delayed cardiovascular failure. As a highly efficacious therapeutic class used in the management of a wide range of cancers, the position of anthracyclines as a mainstay of the chemotherapy artillery cannot be disputed.4 Many approaches to reduce the cardiotoxicity of anthracyclines have been explored and have had varying degrees of success. Encouragingly, studies using prophylactic cardiological therapies, such as angiotensin modulating therapies, have shown significant promise in the clinic.5
Disruption of angiotensin signalling as a therapeutic strategy
Interference with angiotensin signalling cascades, using angiotensin-converting enzyme inhibitors (ACEi) or angiotensin-receptor blockers (ARBs), and the resulting clinical effects on cardioprotection from anthracycline-induced cardiotoxicity strongly supports a relationship between this toxicity and angiotensin signalling.6–9 Despite this, the mechanistic basis of this effect has not been reported and as such identification of susceptible patients or risk mitigation strategies remain to be developed. To uncover these molecular mechanisms, I utilised in vitro cardiomyocyte models paired with impedance-based technologies to investigate the effects of ACEi or ARBs on mitigation of anthracycline-induced cardiotoxicity.
The structural changes are pivotal
Chronic anthracycline-induced cardiotoxicity is associated with structural changes to cells which reduce cardiac function and can lead to heart failure. This is especially pertinent in survivors of childhood cancer where deterioration of cardiac function can arise decades after treatment following a long asymptomatic period.10 Interestingly, one of the effects of angiotensin signalling on the cardiac system is cardiac hypertrophy and remodeling,11 which suggests a crossover of physiological outcomes between angiotensin signalling and anthracycline-induced cardiotoxicity. Following qualification of the cardiomyocyte models for detection of structural changes using the hypertrophic mediator angiotensin II and the structural cardiotoxicant sunitinib, an investigation of the effects of anthracyclines with and without ARBs on cell morphology was conducted. This demonstrated that clinically relevant doses of anthracyclines (50-500nM) induced cellular hypertrophy, an adverse effect that was mitigated by concomitant ARB treatment. In the cardiomyocyte cell line model, the reduction in hypertrophy induced by ARB treatment was paired with increases in cell viability, providing further evidence that blockade of the ATR1 is cardioprotective. Importantly, no concomitant reduction of anthracycline efficacy against tumour cells was detected, an important consideration for clinical translation of this study. Therefore, the reduction in cardiomyocyte hypertrophy observed in this study supports blockade of angiotensin-receptor 1 (ATR1) as at least partially responsible for the protection afforded by ARBs against anthracycline-induced cardiotoxicity in patients.
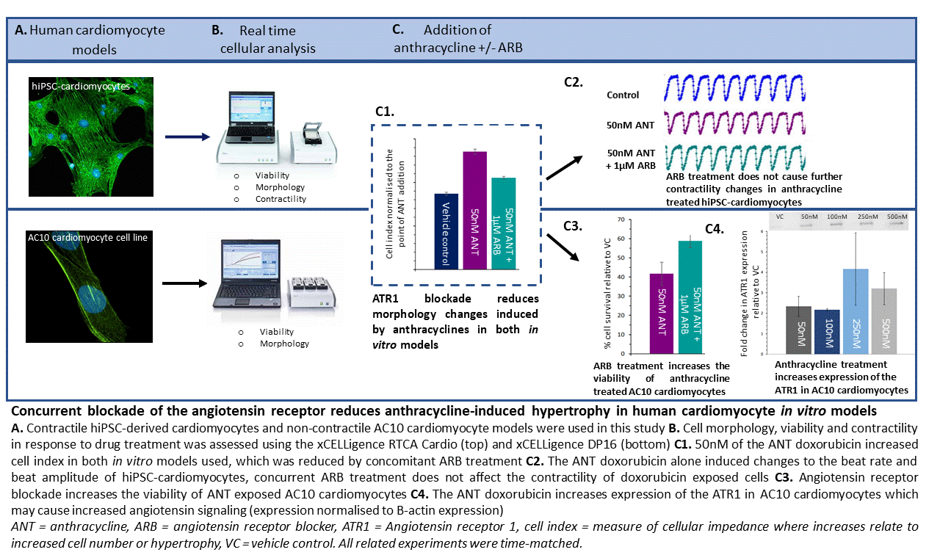
This hypothesis is substantiated by analyses of molecular changes in the in vitro cardiomyocyte cell model after anthracycline exposure, which revealed a 3–4-fold increase in protein expression of ATR1, and increased gene expression of regulatory proteins required for angiotensin signalling. Additionally, I showed anthracycline exposure also increased expression of genes relating to oxidative stress and cardiac remodelling; this is a clear area of overlap between angiotensin signalling and anthracycline toxicity. Activation of these pathways may further potentiate the cardiotoxic effects of anthracyclines and thus begin to explain why reducing angiotensin signalling demonstrates cardioprotective effects both in clinical studies and the present study. Going forward it would be useful to assess effects on other cell types found in the heart, such as cardiac fibroblasts and endothelial cells, following anthracycline treatment to establish the multicellular nature of these cardiac effects.
Translational potential and personalised treatment
The use of cardiomyocytes from patients with and without anthracycline-induced cardiotoxicity offers the potential to build on the current research in a translational manner.12 Studies that evaluate the likelihood of anthracycline-induced hypertrophy occurring and if ARB therapy is effective at reducing hypertrophy in all or just some patient cells would further appraise the use of ARBs as a cardioprotective strategy.
This research also has further clinical implications as the systemic level of angiotensin II is determined by the polymorphic genotype of the angiotensin converting enzyme (ACE), which is responsible for the final step of angiotensin II production. A patient expresses one of three polymorphic variant genotypes associated with an insertion/deletion within an intronic region of this gene: I/I, I/D and D/D, associated with low, medium and high levels of angiotensin II, respectively. Previous studies demonstrate an association between the D/D genotype and increased risk of cardiac failure, resulting from the higher levels of angiotensin II present in the blood.13,14 This risk may extend to anthracycline-induced cardiotoxicity, based on an association between this toxicity and increased angiotensin signalling.
The concept of successful cancer treatment is now extending beyond cancer free survival to include management of long-term effects such as cardiotoxicity, to ultimately improve the quality of life for cancer survivors. The outcome of my study has shown synergism between angiotensin signalling and anthracycline-induced cardiotoxicity, suggesting that a reduction in angiotensin signalling is clinically protective against this toxicity. This increased understanding will pave the way for improved management of anthracycline-induced cardiotoxicity as patient risk can be stratified by identifying biomarkers for high-risk patients, and prophylactic medications to limit the occurrence of the life-threatening cardiotoxicity can be administered as necessary.
References
- Maddams, J., Utley, M. & Møller, H. Projections of cancer prevalence in the United Kingdom, 2010-2040. Br. J. Cancer 107, 1195–202 (2012).
- Ewer, M. S. & Ewer, S. M. Cardiotoxicity of anticancer treatments. Nat. Rev. Cardiol. 12, 547–58 (2015).
- Han, X., Zhou, Y. & Liu, W. Precision cardio-oncology: understanding the cardiotoxicity of cancer therapy. npj Precis. Oncol. 1, 1–11 (2017).
- Volkova, M. & Russell, R. Anthracycline Cardiotoxicity: Prevalence, Pathogenesis and Treatment. Curr. Cardiol. Rev. 7, 214–220 (2012).
- Kalam, K. & Marwick, T. H. Role of cardioprotective therapy for prevention of cardiotoxicity with chemotherapy: A systematic review and meta-analysis. Eur. J. Cancer 49, 2900–2999 (2013).
- Cardinale, D. et al. Prevention of high-dose chemotherapy-induced cardiotoxicity in high-risk patients by angiotensin-converting enzyme inhibition. Circulation 114, 2474–81 (2006).
- Janbabai, G. et al. Effect of Enalapril on Preventing Anthracycline-Induced Cardiomyopathy. Cardiovasc. Toxicol. 17, 130–139 (2017).
- Nakamae, H. et al. Notable effects of angiotensin II receptor blocker, valsartan, on acute cardiotoxic changes after standard chemotherapy with cyclophosphamide, doxorubicin, vincristine, and prednisolone. Cancer 104, 2492–2498 (2005).
- Cadeddu, C. et al. Protective effects of the angiotensin II receptor blocker telmisartan on epirubicin-induced inflammation, oxidative stress, and early ventricular impairment. Am. Heart J. 160, 487.e1–7 (2010).
- Lipshultz, S. E., Alvarez, J. A. & Scully, R. E. Anthracycline associated cardiotoxicity in survivors of childhood cancer. Heart 94, 525–533 (2008).
- Higuchi, S. et al. Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology. Clin. Sci. (Lond). 112, 417–28 (2007).
- Burridge, P. W. et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 22, 547–56 (2016).
- Danser, A. H. et al. Angiotensin-converting enzyme in the human heart. Effect of the deletion/insertion polymorphism. Circulation 92, 1387–8 (1995).
- Cambien, F. et al. Deletion polymorphism in the gene for angiotensin-converting enzyme is a potent risk factor for myocardial infarction. Nature 359, 641–4 (1992).




